
我院萬穎教授團隊利用Pd基間隙固溶體催化劑實現了均相體系中難以發生的一個新反應:五元雜環化合物高選擇性直接雙芳基化反應。研究成果以“Unleash electron transfer in C-H functionalization by mesoporous carbon supported palladium interstitial catalysts”為題,在線發表于《國家科學評論》(National Science Review),第一作者為趙小瑞,通訊作者為萬穎教授,通訊單位是上海師範大學。

“芳環-雜芳環-芳環”結構是生物活性分子、藥物和光電材料中的重要結構單元。怎樣的催化劑可以催化這類分子的合成呢?
均相金屬催化劑往往隻能對雜環分子的單一位點進行芳基化;傳統的工業多相催化劑(例如Pd/C)更是連雜環分子單芳基化反應都難以發生。
這主要是因為:(1)N或S與金屬的配位作用導緻雜環分子定向轉化可控性差;(2)含N或S的雜環結構在Pd表面容易發生強化學吸附,導緻催化劑流失或中毒。因此,提高雜環分子定向轉化的選擇性和催化劑穩定性是一個艱巨的挑戰。
萬穎教授課題組在前期研究中發現:(1)介孔炭載Pd基間隙固溶體催化劑可有效調控Pd位點電子結構,有望調節雜環分子的吸附構型和吸附能(ACS Catal. 2017, 7: 2074–2087);(2)利用實驗測量的Pd位點d帶電荷密度,可以線性關聯複雜反應物吸附生成活性絡合物的吸附活化熵(ΔS0*)和轉化頻率(TOF)(Nat. Commun. 2019, 10: 1428)。
在此基礎上,研究者提出,可以基于實驗測量的Pd位點d帶電荷密度,重新設計催化劑,降低強吸附質在Pd位點的吸附能,變革傳統合成路線,構建雜環分子直接雙芳基化新反應(如下圖)。
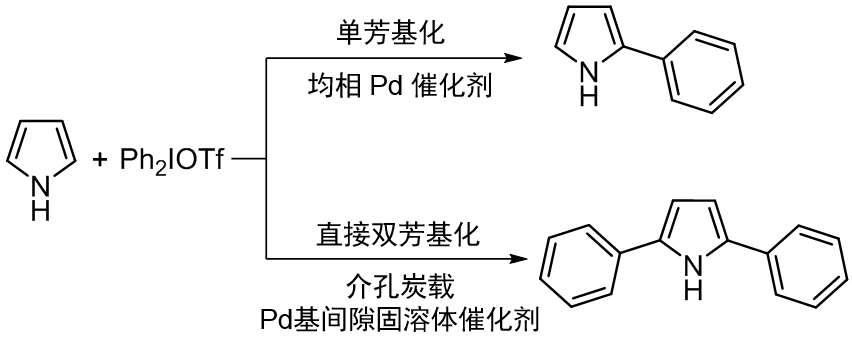
研究者設計了含C和N的Pd基間隙固溶體催化劑,提高Pd位點d帶電荷密度,并通過多位點電子轉移,構建了吡咯及衍生物C-H鍵直接雙芳基化新反應,提高催化劑活性和選擇性,一步獲得2,5-二苯基吡咯等。
這一反應突破了傳統均相催化劑單位點電子轉移一步僅能獲得單芳基産物的限制,為反應物C2/C5位創造了雙親電位點,實現了C-H鍵直接雙功能化反應;同時有效解決了催化劑流失和中毒失活等問題。新的多位點電子轉移精準構建C-C鍵反應路徑,可用于呋喃、噻吩等雜環分子直接雙芳基化反應中,一步獲得2,5-二苯基呋喃、2,5-二苯基噻吩等産物(如下圖)。

這些結果說明,基于多相催化劑表面多位點吸附活化,甚至可以實現均相體系中難以發生的催化過程。